We maintain a variety of specialised, freely-available, open-source informatics tools that help research scientists from around the world answer specific research questions and identify and prioritise potential therapeutic drug targets.
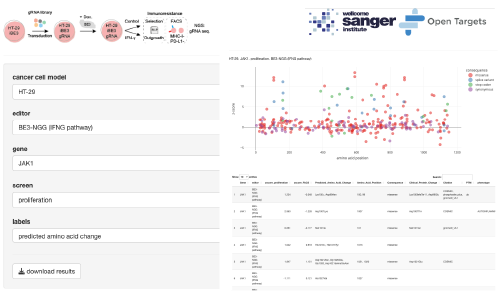
BE-view
BE-view is an R Shiny app. Interact with data from base editing mutagenesis screens to understand the functional consequence of variants in the IFNg signalling pathway. For more information, read the publication (Coelho et al. 2023).
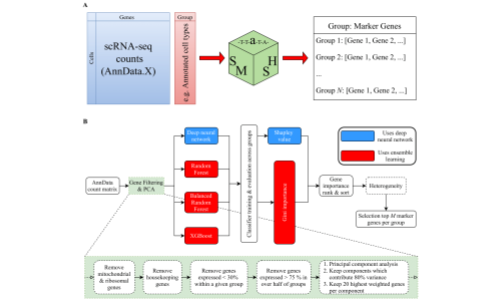
SMaSH
The SMaSH (Scalable Marker gene Signal Hunter) framework is a general, scalable codebase for calculating marker genes from single-cell RNA-sequencing data for a variety of different cell annotations as provided by the user, using supervised machine learning approaches. For more information, see Nelson et al., 2021.
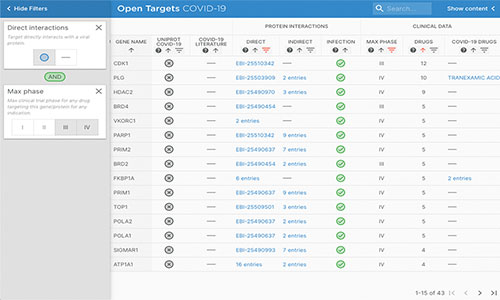
COVID-19 Target Prioritisation Tool
The COVID-19 Target Prioritisation Tool (now deprecated) was an interactive, open source web portal that integrated molecular and clinical data from EMBL-EBI and other public resources to provide an evidence-based framework to support decision-making on potential drug targets and treatments for COVID-19. It has now been deprecated. For more information, see our release blog post.
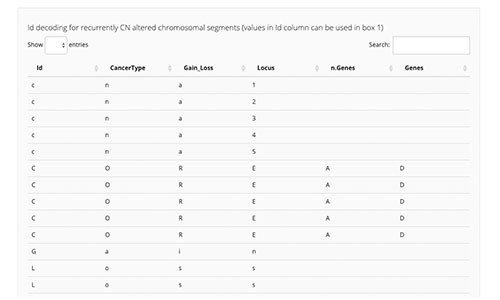
CELLector
CELLector is an open source R tool and package that leverages tumour genomics data to explore, rank, and select optimal cell line models, enabling scientists to make appropriate and informed choices about model inclusion and exclusion in retrospective analyses and future studies. For more information, see Najgebauer et al. 2020.
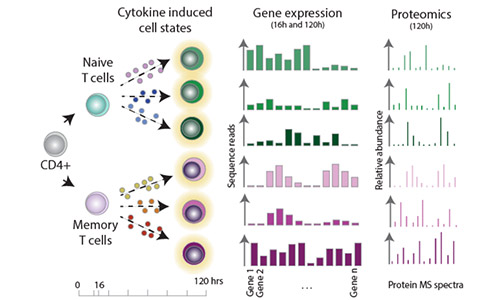
Effectorness
Naïve CD4+ T cells coordinate the immune response by acquiring an effector phenotype in response to cytokines. Here, we used quantitative proteomics, bulk RNA-seq, and single-cell RNA-seq to generate a detailed map of cytokine-regulated gene expression programs. For more information, see Cano Gamez et al. 2020 and our Effectorness project page.
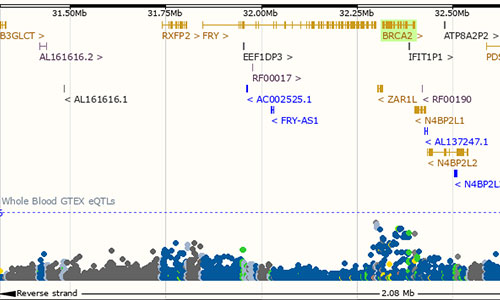
eQTL Catalogue
The eQTL Catalogue provides uniformly processed gene expression and splicing QTLs from all available public studies on human. This resource focuses on expression cis-QTLs where variants are associated to expression levels of nearby genes, and on splicing QTLs where variants are associated to specific splicing events on nearby splice junctions.
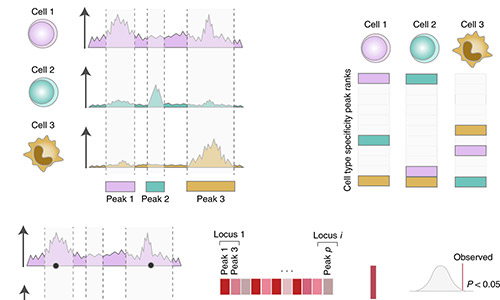
CHEERS
CHEERS (Chromatin Element Enrichment Ranking by Specificity) is a computational pipeline that determines enrichment of annotations in GWAS significant loci. In addition to SNP-peak overlap, CHEERS takes into account peak properties as reflected by quantitative changes in read counts within peaks. For more information, see Soskic et al. 2019.
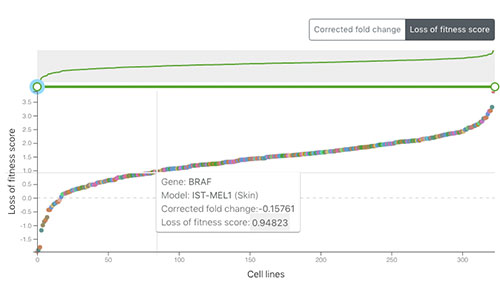
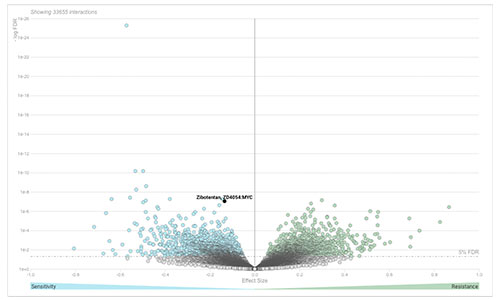
Project Score
Project Score is a web portal that allows researchers to explore the results of CRISPR-Cas9 whole-genome drop out screens across a diverse collection of human cancer cell models and to identify dependencies in cancer cells to help guide early-stage drug discovery for precision cancer medicines. For more information, see Behan et al. 2019.
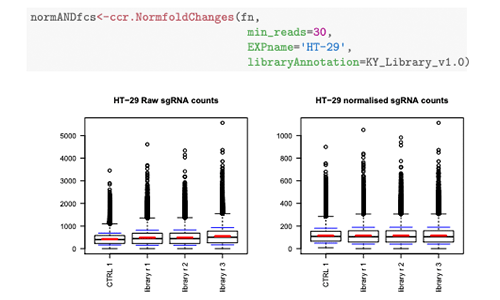
CRISPRcleanR
CRISPRcleanR is a computational tool that identifies and corrects gene-independent responses to CRISPR-Cas9 targeting. CRISPRcleanR is available as an R package, as a Python package, and as a dockerised, platform-independent Python implementation suitable for cloud environments. For more information, see see Iorio et al. 2018.
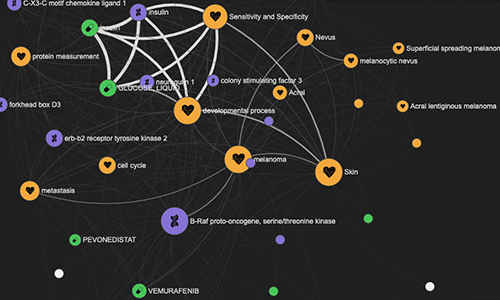
DoRothEA
DoRothEA (Discriminant Regulon Expression Analysis) is a web tool that can be used to search candidate TF-drug interactions in cancer. We studied the role of 127 TFs in drug sensitivity across more than 1,000 cancer cell lines screened with 265 anti-cancer compounds from the GDSC. For more information, see Garcia-Alonso et al. 2017
LINK
LINK (LIterature coNcept Knowledgebase) is a now-deprecated pipeline and web tool that allowed for the exploration of half a billion relations between genes, diseases, drugs, and key concepts extracted from PubMed abstracts using natural language processing. With the launch of the next-gen Platform in April 2021, we replaced LINK with a new Bibliography pipeline.
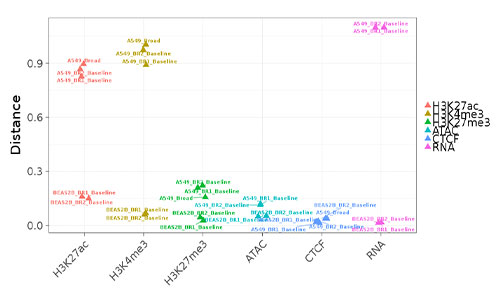
epiChoose
epiChoose is a now-deprecated tool for quantifying the relatedness between cell lines and primary cells. We extensively profiled a number of commonly used cell line models across a number of tissues. This profiling consists of epigenetic (histone modification, CTCF, ATAC-seq) and transcriptional (RNA-seq) whole-genome measurements.